- ITソリューショントップ
-
製品・ソリューション
-
ダイキンのIT
 製造業向けITソリューション
製造業向けITソリューション 品質DX支援 QX digital solution
品質DX支援 QX digital solution 建設業務改善ソリューション
建設業務改善ソリューション ビル管理業務支援 DK-CONNECT BM
ビル管理業務支援 DK-CONNECT BM FILDER SiX TOP
FILDER SiX TOP FILDER SiX 電気 TOP
FILDER SiX 電気 TOP Rebro D TOP
Rebro D TOP 実験記録をデータベース化 ParsleyLab
実験記録をデータベース化 ParsleyLab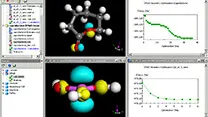 マテリアルサイエンス向けソフト Materials Studio
マテリアルサイエンス向けソフト Materials Studio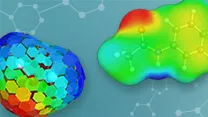 熱力学物性予測ソフトウェア COSMO
熱力学物性予測ソフトウェア COSMO 電子実験ノート
電子実験ノート 総合3DCG 制作ソフトウェア Maya
総合3DCG 制作ソフトウェア Maya 総合3DCG 制作ソフトウェア 3ds Max
総合3DCG 制作ソフトウェア 3ds Max 3Dキャラクタアニメーション制作ソフトウェア MotionBuilder
3Dキャラクタアニメーション制作ソフトウェア MotionBuilder モーションキャプチャーシステム Xsens MVN
モーションキャプチャーシステム Xsens MVN
分子モデリング・
シミュレーションソフトウェア
Materials Studio
Materials Studio 研究分野別パッケージ
触媒
お勧めパッケージ
Adsorption Solver Pack+Base もしくは DMOL3 Solver Pack+Base
このような用途にお勧めします。
- 触媒のメカニズムを分子レベルで解明したい。
- 分子レベルで、どの場所に吸着しているかを予測したい。
パッケージ構成
Adsorption Solver Pack
- Adsorption Locator 吸着場所の推定を簡易計算で実施、候補箇所を推定できます。
- Sorption 吸着性シミュレーションプログラム
DMOL3 Solver Pack
- DMol3 第一原理計算による吸着箇所、反応状況の精密計算ができます。
Base
- Materials Visualizer 基本パッケージ、構造の作成、修正、計算結果の表示解析します。
- Gaussian I/F Gaussianの入力ファイル作成、計算結果の読み込みができます。(Gaussian は別途契約が必要です)
- VAMP 半経験的分子軌道計算で、分子構造の最適化や電荷計算が行えます。
適用例
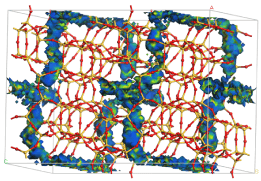
100 kPa、300 K の条件でゼオライトMFI 中のメタンの吸着を表したものです。
CHの予測位置の表面を結合エネルギーで色づけ表示してます。エネルギーの低い(結合力は強い)エリアはレッド、エネルギーの高いエリアはブルーで示されています。
このような図によって、孔の中の優先的結合部位とともに、吸着物が高濃度に存在するエリアが示されます。
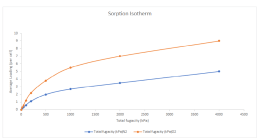
300Kのモルデナイト中の酸素と窒素の収着等温線をMaterials Studio(Sorption)を使った計算結果です。吸着物の混合物を処理する能力は、分離および拡散のプロセスをモデル化する場合に重要です。
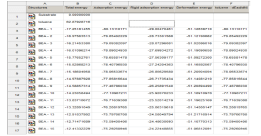
計算から得られた一覧は、幾つかのエネルギー範囲における最も安定な基材-吸着配置を示しており、多くの配置を簡単にモデル化して比較することができます。結果の解析簡単にできます。
高分子
お勧めパッケージ
Classical Simulation Pack+Base もしくは DMOL3 Solver Pack+Base
このような用途にお勧めします。
- 高分子ポリマーの構造に対する物性値の予測をしたい。
- アモルファル構造を作成し、架橋反応のメカニズムを解明したい。
パッケージ構成
Classical Simulation Pack
- Amorphous Cell アモルファス構造の構築ができます。
- Forcite Plus 分子力場計算、分子動力学計算で、構造の安定化計算、動力学計算が可能で、次のような物性値を予測できます。
Forcite Plusで予測できる物性値
・全エネルギー計算、・構造最適化計算、・分子動力学計算、・Anneal計算、・Quench計算、・密度、・凝集エネルギー密度、・溶解度パラメータ、・ガラス転移温度、・ガス分子の拡散係数、・界面の相互作用エネルギー、・接着力、・機械特性、・屈折率(赤外領域)、・結合長、・角度、・二面角分布、・動径分布関数、・せん断粘性率、・熱伝導率、・慣性半径、・熱力学特性(熱膨張係数、比熱など)、・溶媒和自由エネルギー、・基準振動解析、・誘電率
- COMPASS DFTより求めた精度の高い力場です。
- Conformers 有機分子のコンフォメーション解析ができます。
DMOL3 Solver Pack
- DMol3 第一原理計算による吸着箇所、反応状況の精密計算ができます。
Base
- Materials Visualizer 基本パッケージ、構造の作成、修正、計算結果の表示解析します。
- Gaussian I/F Gaussianの入力ファイル作成、計算結果の読み込みができます。(Gaussian は別途契約が必要です)
- VAMP 半経験的分子軌道計算で、分子構造の最適化や電荷計算が行えます。
適用例
分子動力学シミュレーションによるエポキシ樹脂の硬化過程の解析と力学特性の予測
九州大学 山本教授 (2019年1月10日東海シンポジウム講演)
Peel-off simulation(DGEBA/44DDS).
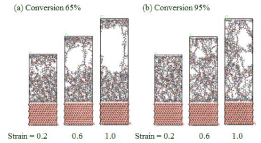
DGEBAについて、反応率65%および95%のときのスナップショットです。フィラーとエポキシ樹脂の界面は歪1まで接着した状態であり、樹脂内部にボイドが形成されて高分子鎖が引き伸ばされている。
Peel-off simulation(DGEBA/44DDS).
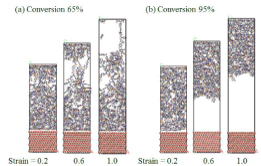
反応率65%ではDGEBAと同様に樹脂内部にボイドを形成し、高分子鎖が引張り方向に引き伸ばされており、界面では樹脂が剥がれておらず、凝集破壊となっている。しかし、反応率95%では界面から剥離が起こっている。
半導体、金属研究
お勧めパッケージ
CASTEP Solver Pack+Base
このような用途にお勧めします。
- 半導体物質のバンド構造を予測、有機分子のNMR予測、金属表面EELSスペクトル予測をしたい。
パッケージ構成
- CASTEP 第一原理計算によるバンド計算、NMR、EELS予測計算ができます。
- NMR CASTEP 第一原理計算によるバンド計算、NMR、EELS予測計算ができます。
- Materials Visualizer 基本パッケージ、構造の作成、修正、計算結果の表示解析します。
- Gaussian I/F Gaussianの入力ファイル作成、計算結果の読み込みができます。(Gaussian は別途契約が必要です)
- VAMP 半経験的分子軌道計算で、分子構造の最適化や電荷計算が行えます。
CASTEPで計算できる項目
・電子エネルギー、電子密度 ・構造最適化 ・量子分子動力学計算 ・状態密度、エネルギー分散(バンド図) ・フォノン計算(状態密度、バンド) ・IRスペクトル、ラマンスペクトル、分極率 ・分子軌道の可視化 ・Mulliken解析 ・内殻励起スペクトル(XANES, ELNES)
適用例
- ZnOのバンド構造と状態密度分布 (DOS)
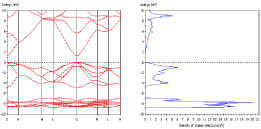
- ELNESスペクトル(CASTEP)
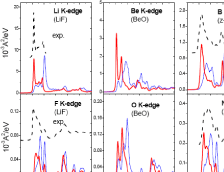
- SnO2のNMR等方遮蔽定数(CASTEP)
X線(有機結晶)
お勧めパッケージ
Powder Diffraction Solver Pack+Base
このような用途にお勧めします。
- 分子構造から、リートベルト解析をしたい。
- 粉末X線の実験データと、分子構造から、結晶パッキング状態の予測をしたい。
パッケージ構成
- ・Reflex 分子構造からリートベルト解析ができます。
- Reflex Plus 粉末X線実験データーと、分子構造から、結晶構造を予測できます。
- Materials Visualizer 基本パッケージ、構造の作成、修正、計算結果の表示解析します。
- Gaussian I/F Gaussianの入力ファイル作成、計算結果の読み込みができます。(Gaussian は別途契約が必要です)
- VAMP 半経験的分子軌道計算で、分子構造の最適化や電荷計算が行えます。
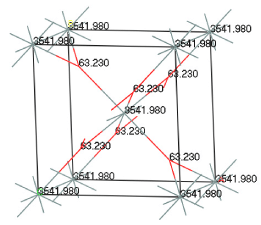
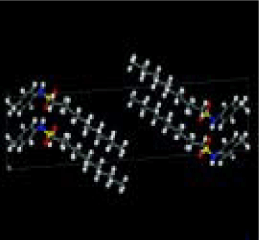
適用例
Reflex Plus を用いて解明されたN-(ρ-tolyl)-dodecyl-sulfonamideの構造
100 kPa、300 K の条件でゼオライトMFI 中のメタンの吸着を表したものです。
CHの予測位置の表面を結合エネルギーで色づけ表示してます。エネルギーの低い(結合力は強い)エリアはレッド、エネルギーの高いエリアはブルーで示されています。
このような図によって、孔の中の優先的結合部位とともに、吸着物が高濃度に存在するエリアが示されます。
グラフィン
お勧めパッケージ
CASTEP Solver Pack+Base
このような用途にお勧めします。
- 有機半導体の化学状態を計算で求め、実験スペクトルとの比較を行いたい。
パッケージ構成
- CASTEP 第一原理計算によるバンド計算、NMR、EELS予測計算ができます。
- NMR CASTEP 第一原理計算によるバンド計算、NMR、EELS予測計算ができます。
- Materials Visualizer 基本パッケージ、構造の作成、修正、計算結果の表示解析します。
- Gaussian I/F Gaussianの入力ファイル作成、計算結果の読み込みができます。(Gaussian は別途契約が必要です)
- VAMP 半経験的分子軌道計算で、分子構造の最適化や電荷計算が行えます。
CASTEPで計算できる項目
・電子エネルギー、電子密度 ・構造最適化 ・量子分子動力学計算 ・状態密度、エネルギー分散(バンド図) ・フォノン計算(状態密度、バンド) ・IRスペクトル、ラマンスペクトル、分極率 ・分子軌道の可視化 ・Mulliken解析 ・内殻励起スペクトル(XANES, ELNES)
計算化学
お勧めパッケージ
Classical Simulation Pack+Base
このような用途にお勧めします。
- 計算化学を行う前の初期構造の構築を簡単に実施。アモルファス構造、ミラー面を指定し、界面構造、周期構造の作成、計算結果の表示が可能です。
パッケージ構成
- Amorphous Cell アモルファス構造の構築ができます。
- Forcite Plus 分子力場計算、分子動力学計算で、構造の安定化計算、動力学計算が可能で、次のような物性値を予測できます。
Forcite Plusで予測できる物性値
・全エネルギー計算、・構造最適化計算、・分子動力学計算、・Anneal計算、・Quench計算、・密度、・凝集エネルギー密度、・溶解度パラメータ、・ガラス転移温度、・ガス分子の拡散係数、・界面の相互作用エネルギー、・接着力、・機械特性、・屈折率(赤外領域)、・結合長、・角度、・二面角分布、・動径分布関数、・せん断粘性率、・熱伝導率、・慣性半径、・熱力学特性(熱膨張係数、比熱など)、・溶媒和自由エネルギー、・基準振動解析、・誘電率
- COMPASS DFTより求めた精度の高い力場です。
- Conformers 有機分子のコンフォメーション解析ができます。
- Materials Visualizer 基本パッケージ、構造の作成、修正、計算結果の表示解析します。
- Gaussian I/F Gaussianの入力ファイル作成、計算結果の読み込みができます。(Gaussian は別途契約が必要です)
- VAMP 半経験的分子軌道計算で、分子構造の最適化や電荷計算が行えます。
オプション
上記のパッケージに自由に組み合わせて追加できます。
| 触媒 | 高分子 | 半導体・金属 | X線(有機結晶) | グラフィン | 計算化学 | |
|---|---|---|---|---|---|---|
| ONETEP Solver Pack | 〇 | 〇 | 〇 | |||
| DFTB+ | 〇 | 〇 | 〇 | 〇 | 〇 | |
| QMERA Solver Pack | 〇 | 〇 | 〇 | 〇 | ||
| Soft Matter Solver Pack | 〇 | |||||
| Mesodyn Solver Pack | 〇 | |||||
| Kinetix | 〇 | 〇 | ||||
| CANTERA | 〇 | 〇 | ||||
| Crystal Morphology Server Pack | 〇 | 〇 | ||||
| FlexTS | 〇 | 〇 |
ONETEP Solver Pack
パッケージ構成
- ONETEP 大規模系(500原子以上) 第一原理計算(DFT)ができます。*並列コンピュータが必要です。
ONETEPで計算できる項目
・単一点のエネルギー計算、・構造最適化、・遷移状態探索、・電子密度、・静電ポテンシャル、・Mulliken電荷、・Mullikenスピン、・結合電子数、・状態密度(DOS)、・分子軌道(MO)
DFTB+
パッケージ構成
- DFTB+ 第一原理計算の精度で、半経験的手法の計算ができます。
DFTB+で計算できる項目
・エネルギー計算、・構造最適化、・NVE,NVT,NPH,NPTアンサンブルを使った分子動力学計算、・パラメータ作成タスク、・力学的特性の計算 ※電子輸送特性の計算(透過関数、I-V曲線)
・非周期系と周期系に適用可能です、・物性(バンド構造、状態密度、電子、スピン、差電子密度、フェルミ面、振動数、波動関数、Mulliken電荷)
Soft Matter Solver Pack
パッケージ構成 ※Mesositeをご利用したい方向けソフトウェア構成
CANTERA
パッケージ構成
- CANTERA 反応速度の計算ができます。詳細な反応機構のデータを基に化学反応や燃焼系のモデル計算を行います。Materials Studioの中でDMol3やCASTEPを用いた第一原理計算とCanteraの反応速度論に基づく計算をシームレスにつなげることができます。
Crystal Morphology Server Pack
パッケージ構成
- Morphology 分子構造から、結晶形状の予測計算ができます。
- Polymorph Predictor 化合物の分子構造から直接その結晶多形を予測計算ができます。
- Motif 分子結晶における水素結合トポロジの情報を分析できます。
FlexTS
パッケージ構成
- FlexTS 化学反応プロセスのシミュレーションにつなげるための従来よりも安定した手法による遷移状態探索ができます。(2021年新機能)
お気軽にお問い合わせください
 電話でお問い合わせ
電話でお問い合わせ
- 東京(担当:SATグループ)
- 03-3520-3082
受付時間 9:00-17:30(土・日・祝除く)