- ITソリューショントップ
-
製品・ソリューション
-
ダイキンのIT
 製造業向けITソリューション
製造業向けITソリューション 品質DX支援 QX digital solution
品質DX支援 QX digital solution 建設業務改善ソリューション
建設業務改善ソリューション ビル管理業務支援 DK-CONNECT BM
ビル管理業務支援 DK-CONNECT BM FILDER SiX TOP
FILDER SiX TOP FILDER SiX 電気 TOP
FILDER SiX 電気 TOP Rebro D TOP
Rebro D TOP 実験記録をデータベース化 ParsleyLab
実験記録をデータベース化 ParsleyLab マテリアルサイエンス向けソフト Materials Studio
マテリアルサイエンス向けソフト Materials Studio 熱力学物性予測ソフトウェア COSMO
熱力学物性予測ソフトウェア COSMO 電子実験ノート
電子実験ノート 総合3DCG 制作ソフトウェア Maya
総合3DCG 制作ソフトウェア Maya 総合3DCG 制作ソフトウェア 3ds Max
総合3DCG 制作ソフトウェア 3ds Max 3Dキャラクタアニメーション制作ソフトウェア MotionBuilder
3Dキャラクタアニメーション制作ソフトウェア MotionBuilder モーションキャプチャーシステム Xsens MVN
モーションキャプチャーシステム Xsens MVN
分子モデリング・
シミュレーションソフトウェア
Materials Studio
CASTEP
密度汎関数理論(DFT)に基づいたab initio(第一原理)量子力学プログラム
CASTEPは密度汎関数法(DFT)によるab initio量子力学計算プログラムです。セラミックス、半導体、金属を含む広範囲な材料の固体、界面、表面の物性をシミュレートするものです。第一原理計算では、構成原子の原子番号以外には何ら実験による情報は必要なく、系の電子的、光学的、構造的特性とその起源を探ることができます。
セラミックス、半導体、金属、界面および表面の性質を計算します。初期構造として与えられた仮想構造から、格子定数、分子の幾何構造、弾性定数、バンド構造、状態密度等の性質予測も可能。CASTEPの活用で、高額な実験の節約や開発サイクルの短縮なども望めます。
CASTEPは、第一原理計算を利用することで、実験に基づいたパラメータを入力することなく、系の電子、光学、および構造の特性に関する本質と起源を調べることができます。経験パラメータや、実験データに乏しい固体物理学、物質科学、および化学工学の分野で起こる課題 の研究に非常に適しています。
CASTEPでは、物質の構造や多くの基本特性を予測することができます。特に、電子特性(バンドギャップやショットキー障壁など)、光学特性(フォノン分散曲線、分極率、誘電率など)、または物理特性(弾性定数など)を予測することができます。insilicoで新し い物質を速く正確に設計するために、すべてを1つのツールにまとめています。主要機能の一つとして遷移状態の探索アルゴリズムがあります。このアルゴリズムを利用すると、反応の理解に必要な反応プロファイルおよびエネルギー障壁の決定に大いに役立ちます。
特徴
周期的構造に対して、弾性係数の6x6のフルテンソルを予測
- 最近のフォノン振動の計算性能向上により、物質の自由エネルギーや熱容量など、熱力学特性の予測が可能になりました。
- 固体系の熱力学的な予測能力により、構造的変化の相安定性など、多くの擬縮物質特性のシミュレーションを実行できます。
格子定数、分子構造、弾性定数、バンド構造、状態密度、電荷密度と波動関数、光学特性などの特性を予測
- 全エネルギー擬ポテンシャル法に基づいて、初期構造として与えられた仮想構造から、格子定数、分子構造、弾性定数、バンド構造、状態密度、電荷密度と波動関数、光学特性などの特性を予測。
- CASTEPの基本となる擬ポテンシャル平面波法は非常に有効であり、毎年出版される何百もの科学的刊行物に、コードを用いた新しい研究成果が掲載されます。このコードの並列版は、数百原子からなる大規模系でも有効です。
適用分野
主な機能
- スピン分極を含めたLDA/GGA計算
- 分子構造、格子定数の最適化
- 全エネルギー、力とストレスの計算
- 幾何構造の緩和内部と外部拘束の有無
- NVEとNVTを使ったMDとLangevin Dynamics
- UV/VISスペクトラ、誘電率
- Mulliken populationと電荷分析
- 結合次数の分析
- 波動関数、電子密度の表示
- バンド構造、状態密度表示
-
詳細機能
- CASTEP TD-DFTの機能で遷移行列の計算が行え、分子の吸収スペクトルの計算が可能
- 計算処理
- 合計エネルギ、力、および圧力
- 構造最適化(単位セル パラメータなど)
- NVE、NVT、NPH、およびNPT集団を使用する分子力学
- 線形および二次的同期トランジット法(LST/QST)に基づいた遷移状態探索
- 弾性定数
- 有限差分あるいは線形応答に基づいたフォノン振動数
- 一般的な機能
- 交換相関相互作用を近似するlocal、gradientcorrected、およびscreened-exchange functionalの選択
- 周期表全体に対するウルトラソフトおよびノルム保存(型)擬ポテンシャル
- ジョブ制御オプション
- 計算効率を最適化する並列化方式の選択
- CPU数の選択
- サーバの指定
- 出力監視とステータスレポート(構造の最適化を行う際のエネルギーと傾きのテキストまたはグラフなど)
- モデルの構造とジョブのステータスをリアルタイムで更新
- Materials Visualizerからリモートサーバのジョブを中止
- 特性
- 光学特性:周波数依存の誘電関数分極率、反射率、屈折率、UVスペクトルなど
- IRスペクトル
- 原子および結合のMulliken population分析
- 静的弾性定数
- フォノン分散
- バンド構造
- 全体および局所フォノン状態密度
- 準調和近似に基づいた熱力学特性(自由エネルギ、エンタルピー、エントロピー、熱容量、Debye温度)
- MS Visualizerを使用したグラフ表示
- 電荷、スピン、および変形密度
- 三次元等高線および二次元スライス
- シミュレートした走査型トンネル顕微鏡(STM)イメージ
- 特性マップによる複数のプロットとカラー面のオーバーレイ
- その他のオプション
- 実空間または逆格子空間の擬ポテンシャル表現
- 空間群の対称性の完全使用
- SCF収束のDIIS、density mixing、smearing、収束を速めるための複数のオプション
CASTEPが使われた論文(敬称略)
- 広島大学 大学院 先進理工系科学研究科 先進理工系科学専攻 松田海斗、准教授 樽谷直紀、教授 犬丸啓
Chemical and Structural Transformations of M–Al–CO3 Layered Double Hydroxides (M = Mg, Zn, or Co, M/Al = 2) at Elevated Temperatures: Quantitative Descriptions and Effect of Divalent Cations - 兵庫県立大学 大学院 工学研究科 応用化学専攻 物質制御計測学研究グループ 教授 村松康司
Correction for the calculated XANES spectra of 1,4,7,10-alkyltetracenes considering the total electron yield (TEY) efficiency of sp2- and sp3-carbon atoms - 兵庫県立大学 大学院 工学研究科 応用化学専攻 物質制御計測学研究グループ 教授 村松康司
Soft X-ray absorption and emission spectra of 2,5,8-triamino-heptazine (melem) with the Theoretical Analysis - 兵庫県立大学 大学院 工学研究科 応用化学専攻 物質制御計測学研究グループ 教授 村松康司
XANESスペクトルデータ(1); 芳香族化合物のCK端XANES - 兵庫県立大学 大学院 工学研究科 応用化学専攻 物質制御計測学研究グループ 教授 村松康司
イミダゾリウム系イオン液体のCK端XANES測定とDFT計算;[CxCy Im]FSAと[CxCy Im]TFSA
事例発表資料
発表資料をご希望の方は下記の資料請求フォームにてご請求ください。
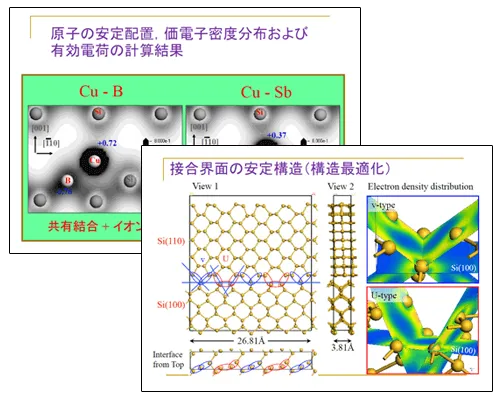
CASTEPを活用した材料開発と材料評価の紹介-半導体シリコン結晶,界面密着性など-
岡山県立大学 情報工学部 末岡浩治氏
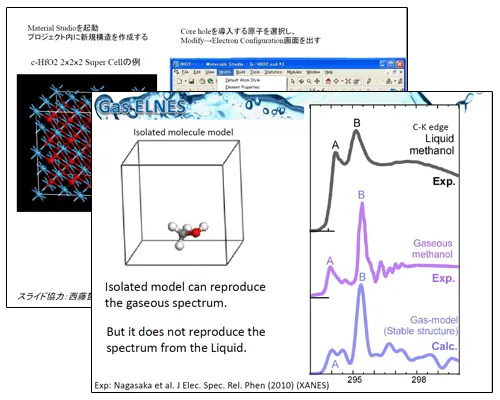
CASTEPを用いたELNES理論計算の基礎と応用
東京大学 生産技術研究所 溝口照康氏
活用分野に最適なMaterials Studio製品をご紹介



お気軽にお問い合わせください
 電話でお問い合わせ
電話でお問い合わせ
- 東京(担当:SATグループ)
- 03-3520-3082
受付時間 9:00-17:30(土・日・祝除く)