- ITソリューショントップ
-
製品・ソリューション
-
ダイキンのIT
 製造業向けITソリューション
製造業向けITソリューション 品質DX支援 QX digital solution
品質DX支援 QX digital solution 建設業務改善ソリューション
建設業務改善ソリューション ビル管理業務支援 DK-CONNECT BM
ビル管理業務支援 DK-CONNECT BM FILDER SiX TOP
FILDER SiX TOP FILDER SiX 電気 TOP
FILDER SiX 電気 TOP Rebro D TOP
Rebro D TOP 実験記録をデータベース化 ParsleyLab
実験記録をデータベース化 ParsleyLab マテリアルサイエンス向けソフト Materials Studio
マテリアルサイエンス向けソフト Materials Studio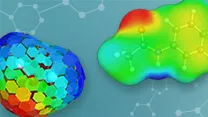 熱力学物性予測ソフトウェア COSMO
熱力学物性予測ソフトウェア COSMO 電子実験ノート
電子実験ノート 総合3DCG 制作ソフトウェア Maya
総合3DCG 制作ソフトウェア Maya 総合3DCG 制作ソフトウェア 3ds Max
総合3DCG 制作ソフトウェア 3ds Max 3Dキャラクタアニメーション制作ソフトウェア MotionBuilder
3Dキャラクタアニメーション制作ソフトウェア MotionBuilder モーションキャプチャーシステム Xsens MVN
モーションキャプチャーシステム Xsens MVN
分子モデリング・
シミュレーションソフトウェア
Materials Studio
より優れた材料でより優れたバッテリーを実現
化石燃料の代替エネルギー源として、多くの用途において、とりわけあらゆる輸送手段を電動化するために、急速充電に対応し、エネルギー密度が高く、安全で長寿命なバッテリーを開発することが喫緊の課題とされています。バッテリー設計は、材料の最小スケール(電子構造)から、バッテリーのセルの構造やパックの設計までのスケールで、熱プロセス、機械的プロセス、化学プロセスの間で起きる複雑な相互作用に依存します。バッテリーやコンデンサーで使用される材料成分を改良することは、必要とされる性能向上を実現するための基礎となります。
Materials Studio には、電解液と電極材料の両方について、重要な材料パラメーター(物性)のシミュレーションを可能にする新機能が用意されています。
1. 新プロトコル:複雑な電解液のイオンと電荷の輸送特性を計算できるように、Materials Studio Collection に新しいプロトコル「Massand Charge Transport」が追加されています。これは、特にバッテリーの電解液成分用に構築されており、リチウムイオンの輸送特性が予測できます。これらの特性は、CATIA Dymola で提供されるような、システムレベルの電池セルモデルのシミュレーションを実行するために必要とされます。これにより、電解液成分を最適化するための条件の一つとして、最終的な電池セルの性能を考慮することができるようになりました。
2. 新プロトコル:Materials Studio Collection に、電極構造へのイオンの挿入から生じる開路電圧を計算するためのもう一つのプロトコルが追加されました。これは、陽極の候補材料(C、Si、Ge など)の安定モデルや複合陰極材料へのリチウムイオンの挿入を想定して構築されていますが、任意のイオンの挿入を扱うことができます。多成分の陰極材料で、それらの正確な構造が未知である場合、クラスター展開法(Materials Studio Collection で使用可能) によって生成される構造を使用することができます。この「Open-Cell Voltage」プロトコルでは、指定したイオンを使用する材料の、ハーフセル(半電池) の電位差が計算できます。このプロトコルでは、全エネルギーの計算に CASTEP が使用され、指定した任意のサーバーで実行できます(複数のサーバーで分散実行が可能)。
金属合金 – 密度汎関数理論から CALPHAD データベースへ
金属合金材料の特性は、材料に含まれるさまざまな原子の安定な結晶相からなる複雑な粒状構造によって決まります。これらの相は、組成、温度、圧力に依存してエネルギーが最低となる構造を反映しており、クラスター展開法と、密度汎関数理論に基づく近似手法により得られる全エネルギーを使用して予測することができます。これらの手法では、複数の金属元素の混合物を正確に表現するための組み合わせ問題を取り扱います。BIOVIA Materials Studio Collection には、すでに、ATATツールを使用してこれらの構造を生成するための便利で幅広いプロトコルのセットが含まれています。バージョン 2020 の新機能では、これらが安定な結晶構造と液相でのシミュレーションのデータを使用して、TDB 形式の CALPHAD データベースのファイルを生成するように拡張されています。考慮に入れる相の範囲を準安定相にまで広げることもできます。
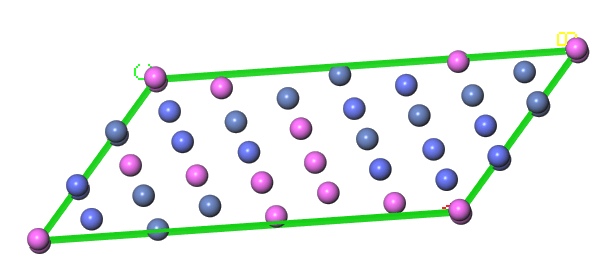
図2:特殊準ランダム構造(SQS)の例:CALPHAD データ作成プロセスの一部分として利用されます。
CALPHAD(Computer Coupling of Phase Diagrams and Thermochemistry)法とそのデータベースは、多成分の相空間でのGibbs 自由エネルギーを評価し、Pandat、ThermoCalc、FactSage などのプログラムを使用して状態図をプロットできる優れた方法となります。
BIOVIA Materials Studio Collection の金属合金用の強力なツールキットを使用することで、バーチャルに設計した金属合金の固化中に生じると考えられる相を予測することが可能になりました。
TBD 形式のファイルを使用して、フェーズフィールド法のシミュレーションコードにおいて、結晶粒や樹枝状結晶自体の成長を正確に表現するために必要な入力を提供することもできます。このように、CALPHAD 法は、この種類の材料の原子スケールとメソ(中間) スケールとの間を橋渡しする重要な技術です。
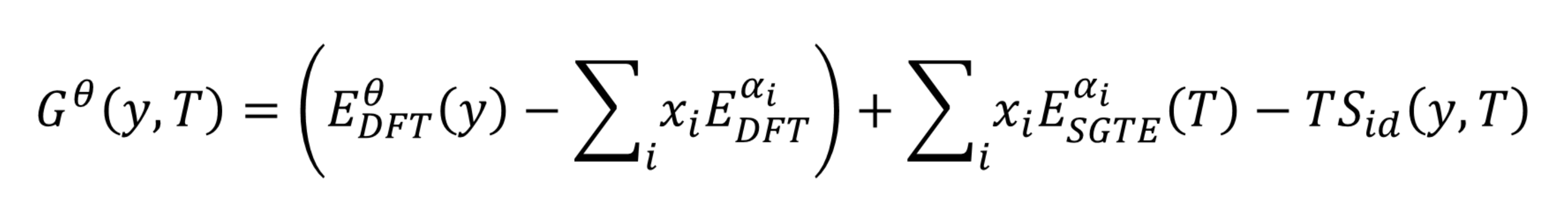
図3:結晶相(ϑ)の関数としての Gibbs 自由エネルギーの式 - CALPHAD データの作成に使用される相ごとに、DFT に基づいて計算された SQS モデルの全エネルギーに対して独立にフィッティングします。
より高い予測精度を実現
古典力学シミュレーション
古典力場に基づくシミュレーションを使用する予測の信頼性は、材料を構成する原子や粗視化粒子(ビーズ)間の相互作用を表現するパラメーターの信頼性に依存します。対象とする材料に適したパラメーターがない場合や、パラメーターがまったくない最悪の状況では、その結果はひどく信頼性に欠けるものになります。BIOVIA Materials Studio Collection 2020 では、そのようなときに、新しい最適化されたパラメーターを作成するためのプロトコルを使用できるようになりました。
新プロトコル:分子内相互作用(結合長、結合角、二面角)のパラメーターを、DMol3 によって計算される全エネルギーと力を再現するように決定する自動処理が、単一の Pipeline Pilot プロトコルに組み込まれています。
新プロトコル:2 つ目のプロトコルは、凝縮相の材料特性を再現するようにファンデルワールス力のパラメーターを決定するために利用できます。
詳細なチュートリアルとして、これらのプロトコルを使って新しいパラメーターを追加するための作業を段階的に実行する例が追加されています。これらは、Pipeline Pilot Connector を介して、Materials Studio から直接実行できます。
![Bis[( trifluoromethyl )sulfonyl]imide](/SC/pics/ms-2020-img04.png)
図4:Bis[( trifluoromethyl )sulfonyl]imide –
リチウム塩となるこのイオン液体分子は一般に普及しているリチウムイオン電池の電解液として使われています。
COMPASS III において、このようなイオン液体向けのパラメーターの開発が行われました。
新パラメーター:BIOVIA Materials Studio COMPASS は、長い間、様々な材料に対して古典力学的シミュレーションを高精度に行うための世界最先端の力場として利用されてきました。Materials Studio 2020 では、新しいプロトコルとして実装された自動化手法を利用して開発を行った、COMPASS 力場の次世代のリリースとなる COMPASS III が追加されています。COMPASS III は、Maybridge,、PoLyInfo、ILThermo の各データベースに登録されている数千に及ぶ一般的な構造に適用できるように開発された、イオン液体を含む広範囲の材料向けの高精度のパラメーターセットです。
量子力学 - DFT の汎関数
CASTEP の新しい汎関数:BIOVIA Materials Studio CASTEP にメタGGA 汎関数の RSCAN が追加されました。これは、汎用的な新しい SCAN 汎関数で、元の定式化におけるいくつかの不安定性が解消されています。RSCAN は、幅広い固体系と分子系にわたって適用でき、かつ高い精度での計算が可能です。これは、有限変位法を使用するフォノン計算や、NMR(J 結合を除く)の計算に使用できます。
DMol3 の汎関数の拡張:DMol3 に、標準的な libXC ライブラリが実装され、幅広い汎関数(ハイブリッドやメタ GGA ハイブリッド汎関数を含む)を利用できるようになりました。GUI から使用できる汎関数に、PBE0、TPSSh、SCAN0、M06、M06-2X が追加されました。これらは、QSAR、QMERA、Materials Studio Collection内で DMol3 を実行する場合にも使用できます。
DFTB+ のパラメーター作成ツール
新スクリプト:Materials Studio の Examples/Scripting フォルダに、DFTB+ のパラメーター作成を自動化するための複数のスクリプトが追加されました。これらを使用して、元素の組み合わせごとの、Slater-Koster ファイルのハミルトニアンおよび重なり行列に関するパラメーターと短距離反発項の両方を作成するプロセスが大幅に簡素化されます。必要な手順を詳細に説明したチュートリアル「Creating Parameters for DFTB+ 」も提供されています。
COSMO-RS COSMOBase

図5:COSMO 表面の3 次元表示。
新機能:COSMOBase に含まれる DMol3-PBE のデータと一貫性がある設定を使用して DMol3 の COSMO を使用する計算を実行するためのオプションが追加されました。Materials Studio を使用してCOSMO ファイルを作成し、それらを COSMOtherm で使用できるようになりました。BIOVIA COSMOtherm では、COSMO-RS 理論に基づき、単一成分液体および混合液体の両方について溶解度、活量係数、相図、蒸気圧、気化熱、相平衡を高精度に計算できます。
大規模な金属系
新機能:BIOVIA Materials Studio ONETEP に、大規模な金属系をオーダー N 法に基づいて高速に計算するための AQUA-FOE (Annealing and QUenching Algorithm for Fermi Operator Expansion)法 が実装されました。新しい触媒材料の開発を目的として、この手法と使用して、大規模な触媒ナノクラスターモデルに対して、数千個の CPU コアによる並列化計算を行った事例が報告されています。
性能と使いやすさ
新機能:Forcite にマルチタイムステップ法が追加され、Ewald 法と PPPM 法を使用した静電相互作用計算のデフォルト設定として新たに採用されました。静電相互作用のうち逆格子空間で扱われる長距離項は、4倍のタイムステップごとに実行されます。この変更が計算されるエネルギーの精度に与える影響は、皆無またはそれに近いレベルですが、マルチコアによる計算では非常に大きな速度向上がもたらされます。
新機能:Materials Studio の Pipeline Pilot Connector を介して計算を実行するときに、以前のプロトコルの設定を再利用して入力パラメーターを設定できるようになりました。
新プロトコル:Materials Studio Collection に「Molecular Dynamics (LAMMPS)」コンポーネントが追加され、BIOVIA Materials Studio から LAMMPSを用いた分子動力学計算を直接実行する機能が追加されました。このコンポーネントを使用するプロトコルの例(Run LAMMPS MD)も提供されています。
お気軽にお問い合わせください
特性と解析
COOP/COHP の解析
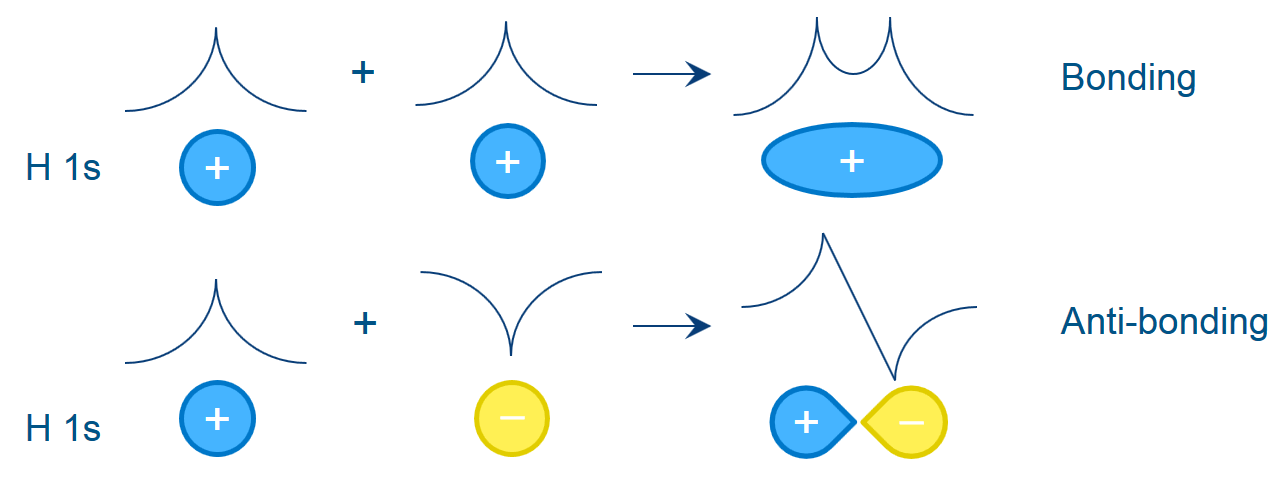
図6:分子軌道が結合性であるか反結合性であるか
を知ることは結合状態を知るうえで非常に重要です。
COOP/COHP の解析では分子だけでなく
結晶についても同様の情報が得られます。
新機能:COOP/COHP(Crystal Orbital Overlap Population/Crystal Orbital Hamiltonian Population)の解析機能が DMol3に追加されました。これにより、分子や結晶の結合状態を解析できるようになりました。COOP/COHP は、軌道エネルギーごとの結合の強さの指標として使用したり、状態密度における特定の状態が結合性であるか反結合性であるかを知るために使用したりすることができます。
Forcite と Mesocite
新機能:Analysis ツールの Concentration Profile の解析では、既存の濃度プロファイルの解析に加えて、質量密度(g/cm3)のプロファイルが出力できるようになりました。
新機能:Analysis ツールの Mean Square Displacement の解析では、統計的に十分なデータがある場合には、平均二乗変位のデータに対して自動的に線形回帰を行うことによって、拡散係数が直接計算されるようになりました。
新機能:Materials Script を介して Forcite の分子動力学計算を実行する際に、振動電場を印加できるようになりました。
新スクリプト:金属結晶の異なる圧力での融解温度を計算するための Z法を実装した Materials Studio スクリプトが利用可能になりました。このスクリプトは Materials Studio の Examples/Scripting フォルダにあります。
MATERIALS STUDIO 2020 に用意されたその他の重要機能
CASTEP
- CASTEP のユーザー・インターフェースが、新しい機能に対応し、使いやすさを向上させるために大幅に変更されました。
- NMR の計算において、G テンソルの計算を行う場合に EPR タスクが使用されるようになり、超微細結合テンソルも計算されるようになりました。超微細結合テンソルの計算に関するドキュメントが、より明確なものになりました。
- CASTEP Analysis ダイアログ IR Spectrum のオプションから、Vibrational Analysis ツールにアクセスできるようになりました。
- ハイスループットスクリーニングなどの目的で使用される OTFG 擬ポテンシャルのライブラリ(QC5)を、MaterialsScript から使用できるようになりました。
- CASTEP のハイブリッド交換相関汎関数を用いた計算で、仕事関数の計算および静電ポテンシャルの可視化ができるようになりました。
GULP
- 分子動力学シミュレーションで温度を柔軟に制御できるように、Anneal タスクが GULP に追加されました。
- GULP がアカデミックバージョン 5.2 に更新されました。機能の主な変更点は以下の通りです。
- - Annealタスクによる分子動力学計算で複数の温度変化を指定可能。
- - (双極子同士が相互作用しない極限での)双極子分極率の計算に 2 次導関数が追加されました。
- - 直方晶系と単斜晶系だけでなく任意の結晶系に対して、領域分割法が利用可能になりました。
- - 微小歪みに対する導関数の計算が可能になりました。
- - イジング模型での結合定数 J2 および J3 に対応するために、原子種の属性としてスピンが追加されました。
- - MEAM に表面エネルギーの計算が追加されました。
- - ReaxFF で、孤立電子対エネルギーおよび過剰配位エネルギーに対する平滑化の処理が改善されました。
- - せん断力を与えるオプションが追加されました。
- - 弾性定数に関する特性の出力が追加されました。
- - Baskes ポテンシャルの二体相互作用項に対して減衰関数が適用できるようになりました。
- - MEAM-2nn-QEq ライブラリが拡張され、Li-Co-O 系と Ti-O/ Si-O 系のパラメーターが追加されました。
ONETEP
- 伝導帯の電子状態の最適化に関連して、局所状態密度(Local DOS)の解析オプションが、ONETEP のユーザー・インターフェースに追加されました。
- 溶媒中でのバルク系と表面系の計算に対応するため、周期的境界条件下で溶媒モデルを使用するオプションが ONETEP に実装されました。
- 大規模な系における結合状態を視覚化する方法として、ONETEP で電子局在関数の解析が可能になりました。
- ONETEP での時間依存密度汎関数理論に基づく励起状態の計算において、以前から使用可能であったノルム保存型擬ポテンシャルに加えて、PAW 擬ポテンシャルを使用した計算が可能になりました。
QMERA
- DMol3 を使用したラマンスペクトルの計算が QMERA に追加されました。
QSAR
- QSAR の VAMP 記述子において、以前から使用可能であった自然原子軌道/点電荷(NAO/PC)モデルでの計算に加えて、ゼロ微分重なり近似(ZDO)を使用して双極子モーメントを計算できるようになりました。
活用分野に最適なMaterials Studio製品をご紹介



お気軽にお問い合わせください
 電話でお問い合わせ
電話でお問い合わせ
- 東京(担当:SATグループ)
- 03-3520-3082
受付時間 9:00-17:30(土・日・祝除く)